



















Challenges In Drug Discovery
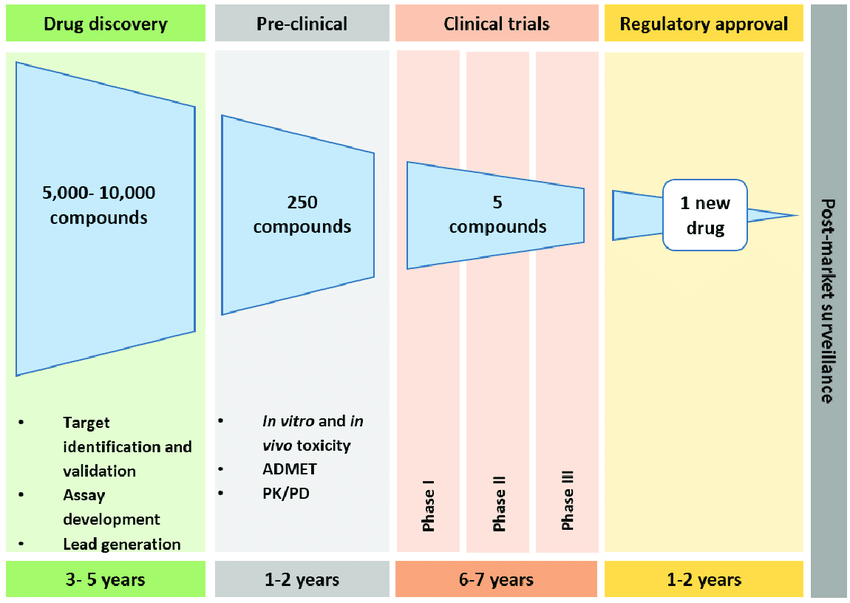
Drug discovery is expensive — both in terms of cost and time. For just one new drug, a biotech company could spend around 3 billion dollars, in a process that can take a decade. The number of permutations of small organic molecules is enormous. Theoretically, there are 10⁶⁵ unique small molecules — so the chemical molecule space is vast [1]! However, not all candidates are easily synthesized. From those “viable” candidates, only a fraction are non-toxic, and then only a fraction of those will have beneficial pharmaceutical properties. Researchers need help finding the “needle in the haystack”.
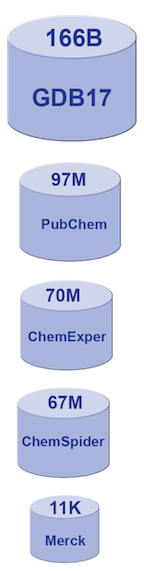
The relative sizes (by number of molecules) of various generally available chemical databases.
Big Data in Bio Pharma
Computational drug discovery is not a new approach to this problem, but the field has seen renewed interest. The general availability of large datasets of chemical compounds has made this possible. Consider the “Merck Index,” the gold standard molecule reference for chemists since 1890. It contains descriptions of 11,000 compounds. There are now molecule databases with sizes into the tens of millions, or more [2,3,4,5,6]. GDB-17, the so-called “chemical universe database”, enumerates 166 billion molecules of up to 17 atoms of C, N, O, S, and halogens. It covers a range containing many drugs that are typical for lead pharmaceutical compounds. Searching large molecule databases for specific pharmaceutical properties can be very challenging.

The Tanimoto similarity metric takes advantage of bit-vectors “finger-prints” of molecules. Each bit explicitly represents the presence or absence of a functional group.
Molecule Search Via Tanimoto Similarity Metric
One search technique involves finding molecules that are structurally similar to molecules with known and desired properties. This is called similarity search and it’s optimized by representing every molecule as a compact “fingerprint.” A common fingerprint format is a simple bit-vector, where each bit position represents the presence or absence of a chemical functional group. A popular scoring metric between two molecules is called Tanimoto. It’s merely the ratio of the number of shared bits divided by the total number present in either molecule. Several recent research papers have shown the effectiveness of the Tanimoto similarity metric [7].
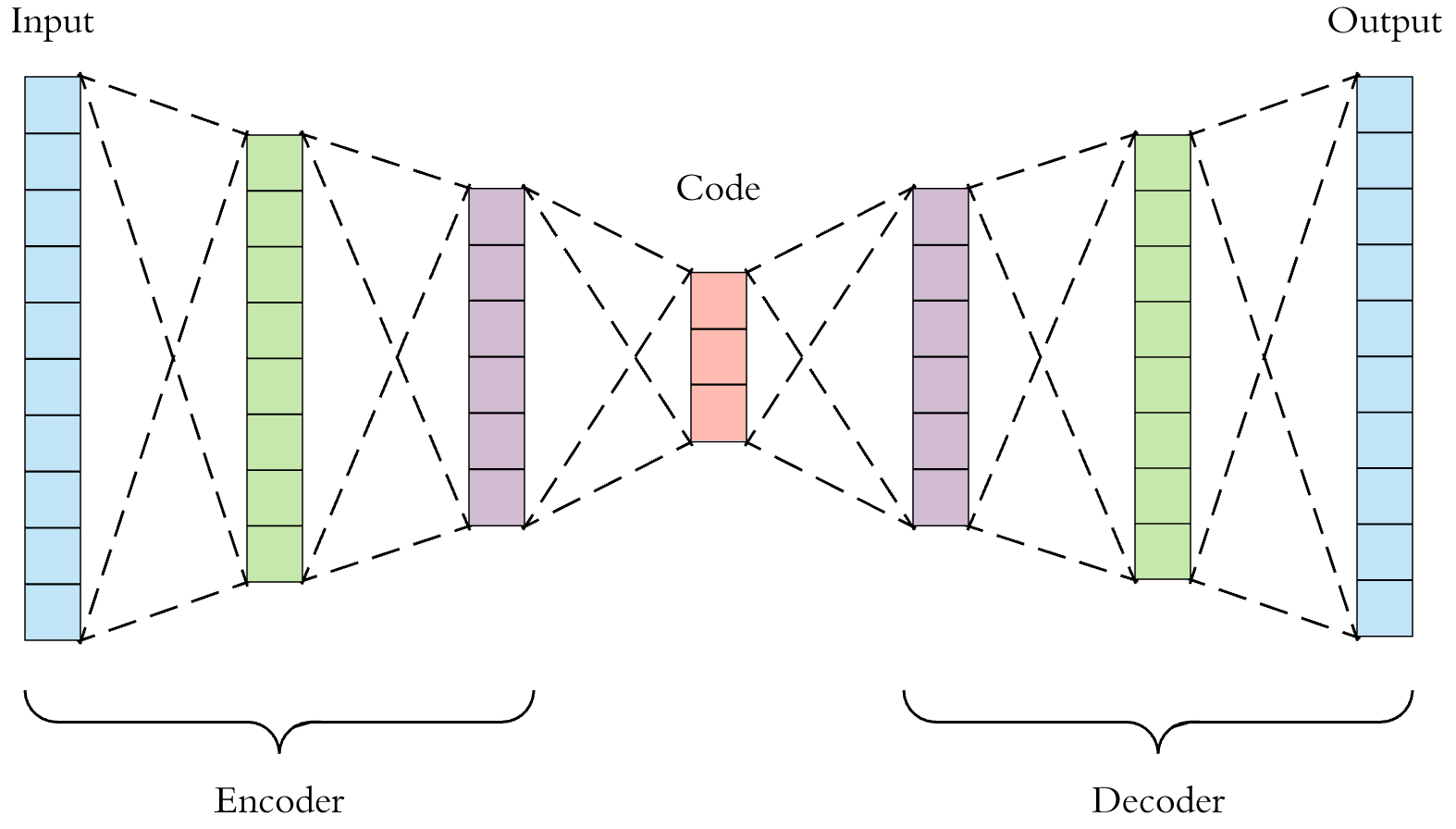
A deep auto-encoder can be used to extract “neural fingerprints” of molecules.
Neural Fingerprints
Some researchers have been applying machine learning to computational drug discovery. Properly trained models can predict molecule properties, and can even generate the blueprints to viable molecules. They can also produce compact fingerprints. For example, the auto-encoder shown above takes raw molecule descriptions and through “self-supervised” training, learns to produce semantically rich numeric codes or “neural fingerprints” at the centermost bottleneck layer. Similarity search performed against a database of neural fingerprints can employ Euclidean, cosine, or hamming distance as a similarity metric. This is a very active area of research, at the intersection of artificial intelligence and cheminformatics [8,9].
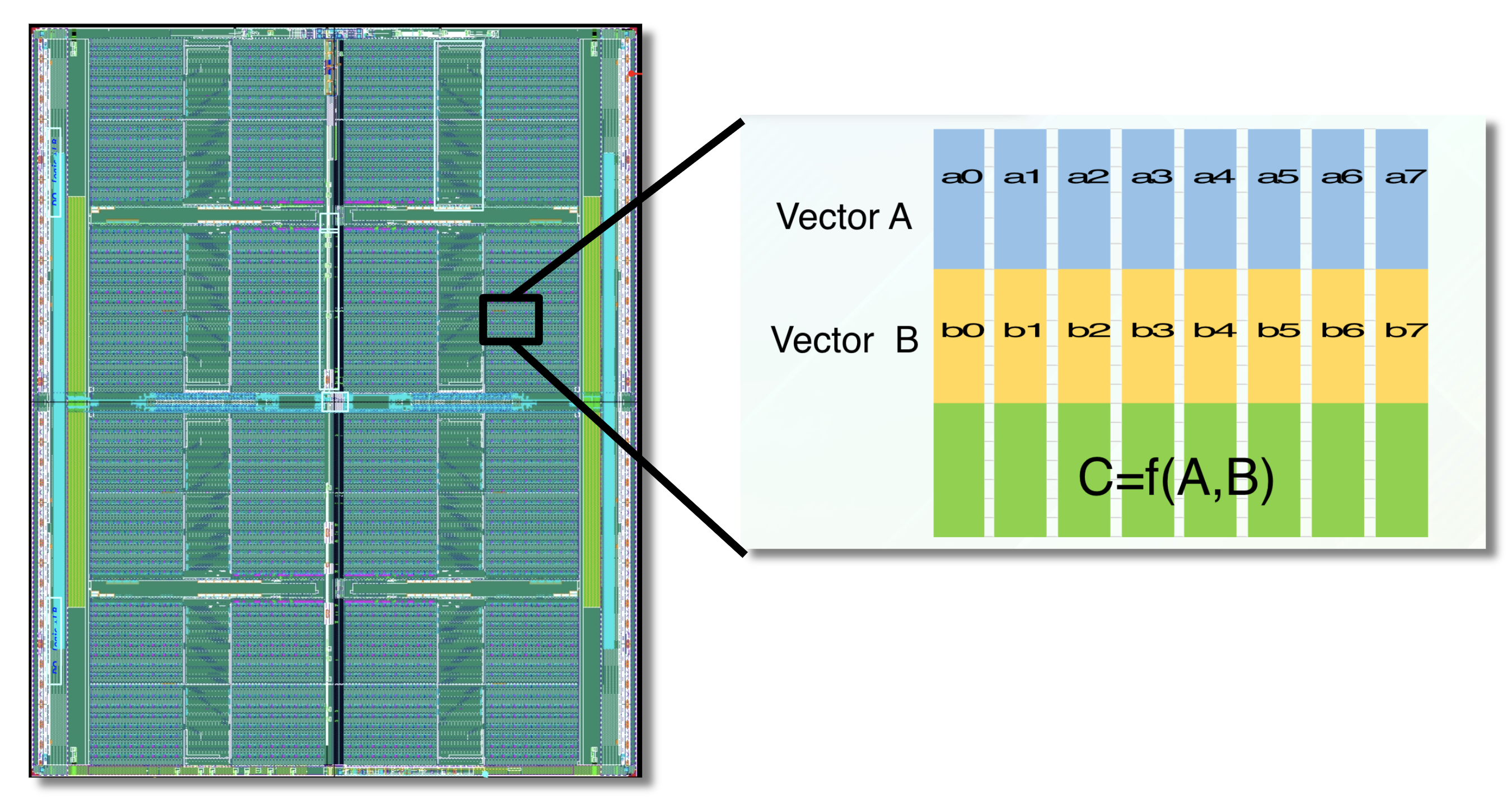
The APU processes data within high-speed SRAM using programmable logic interleaved throughout the chip.
Hardware Acceleration
Custom hardware can accelerate fingerprint-based similarity search. GSI Technology will be introducing the APU (the Associative Processing Unit) for molecule similarity search. The APU is a custom, compute-in-memory SIMD chip, that combines high-speed SRAM and programmable bit-logic, interleaved through-out the memory. It computes functions directly on the data, where the data lives in memory. The APU performs similarity search in a highly parallelized manner, so it’s very fast. Several chips can be combined to enable similarity search on very large molecule databases [10].
Applications
We are integrating our chip with cheminformatics software pipelines, including Dassault’s Biovia Pipeline Pilot [11]. The screen-shots show our custom web-based demo application. Similarity search is as easy as setting a few parameters and choosing a source molecule as the query. Similar molecules are returned instantly.
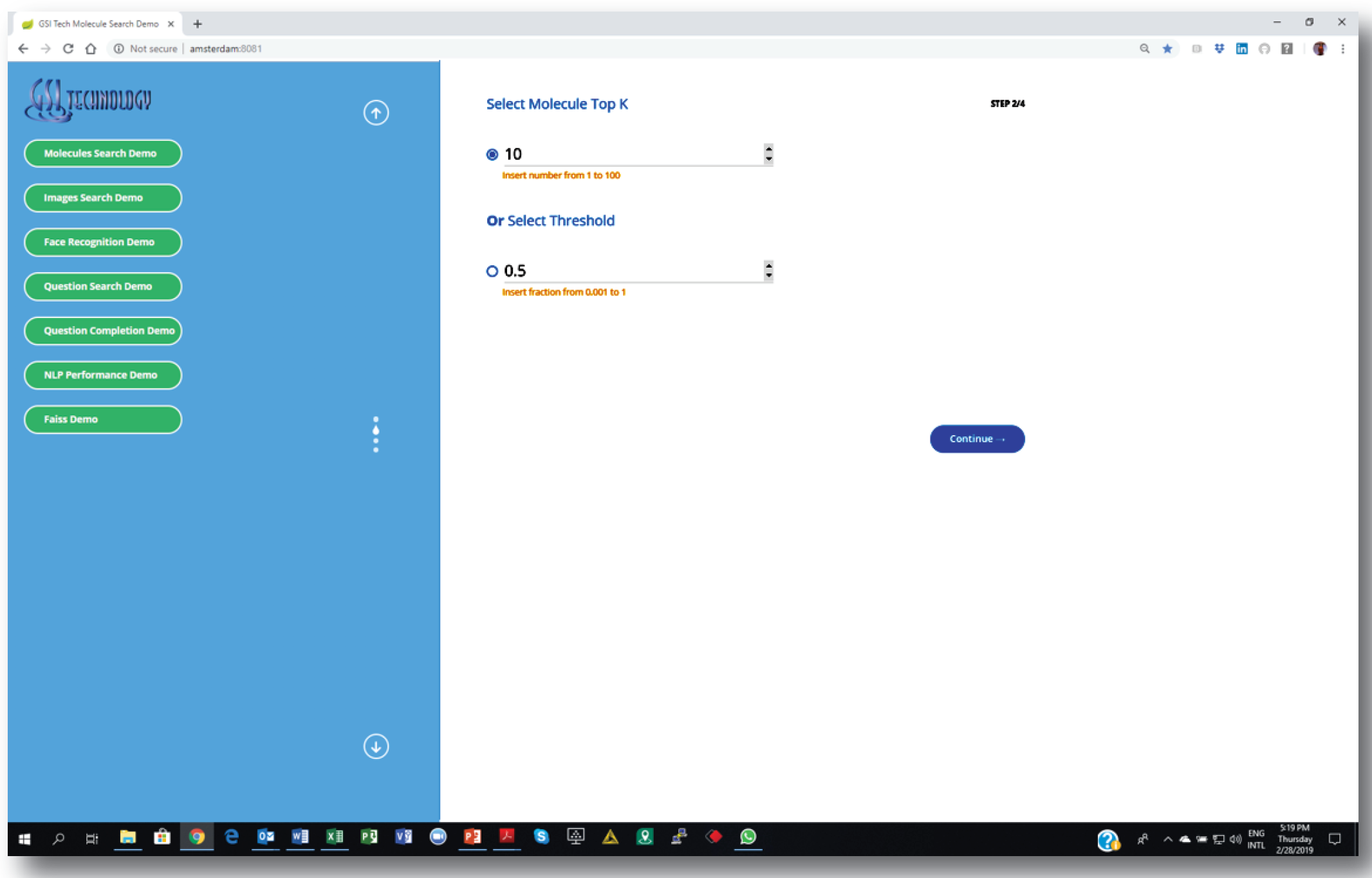
1. Setting similarity search parameters: number of nearest neighbors or “distance” threshold.
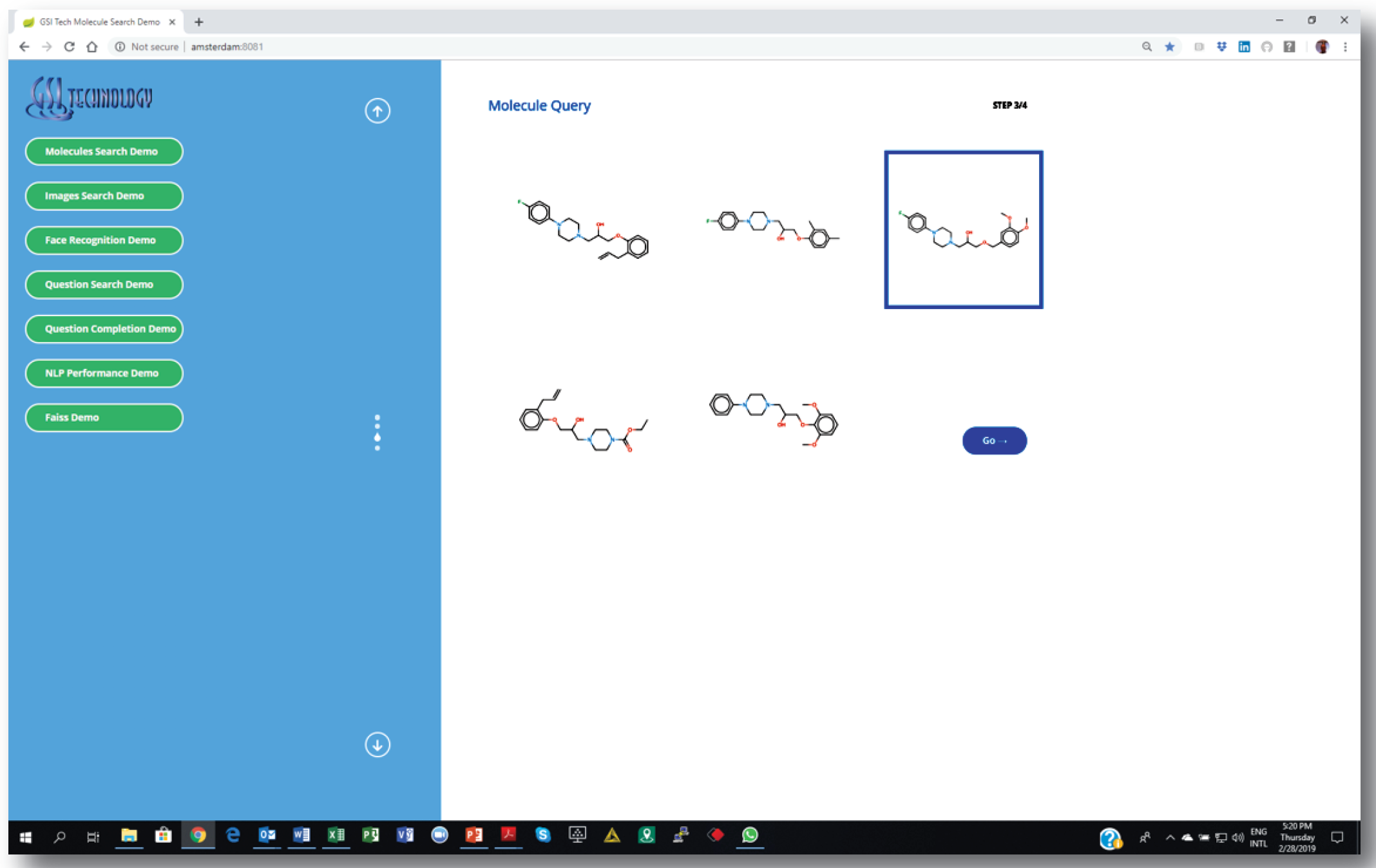
2. Choosing a source molecule for similarity search…
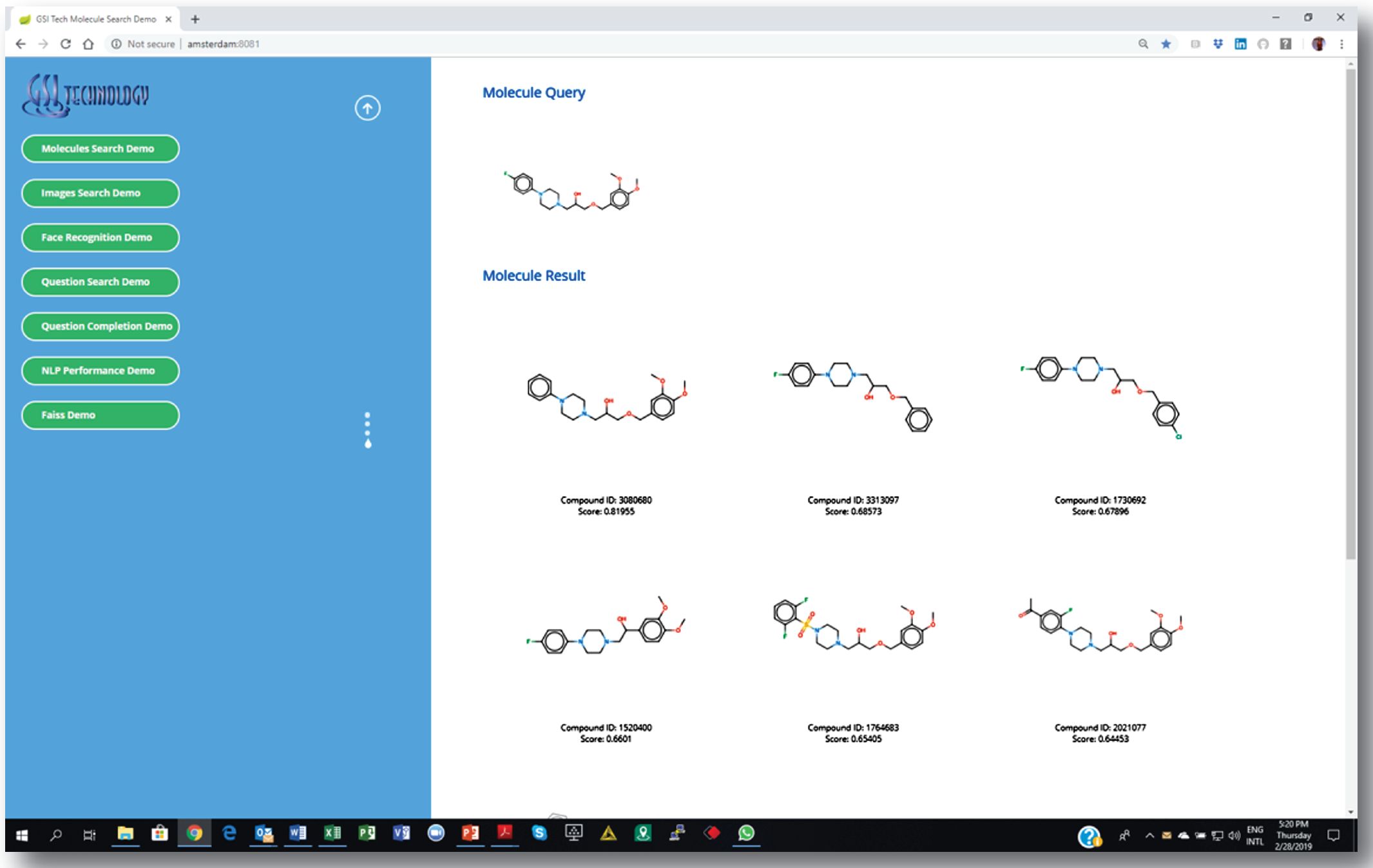
3. Getting a list of similar molecules ranked by a “similarity” score.
The following table shows some performances metrics.
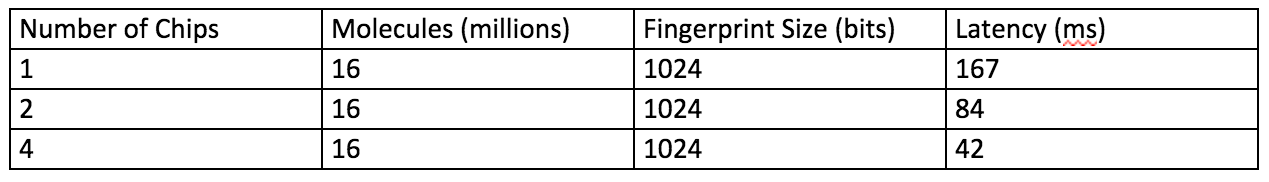
One (1) APU chip can search millions of finger prints with low latency. Multiple chips can support much larger databases.
What’s Next?
We are integrating our chip into existing cheminformatics software applications. We are actively working with major academic and research institutions, enabling hardware-accelerated similarity search on many molecule databases. Hardware-accelerated similarity search is applicable to many tasks, including billion-scale visual search, large-scale face recognition, cybersecurity, and more. Let’s explore the possibilities together. Please reach out to associativecomputing@gsitechnology.com for more information.
References
1. Drug Discovery Timeline Graphic, https://www.researchgate.net/figure/Drug-discovery-and-development-timeline-The-current-drug-approval-pipeline-can-take-15_fig1_308045230
2. Merck Index (on-line), https://www.rsc.org/merck-index
3. ChemSpider (on-line), http://www.chemspider.com/
4. ChemExpr (on-line), https://www.chemexper.com/
5. PubChem (on-line), https://pubchem.ncbi.nlm.nih.gov/
6. “Enumeration of 166 Billion Organic Molecules In The Chemical Universe“, https://pubs.acs.org/doi/10.1021/ci300415d
7. “Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?”, David Bajusz, https://jcheminf.biomedcentral.com/articles/10.1186/s13321-015-0069-3
8. “Automatic Chemical Design Using A Data-Driven Continuous Representation of Molecules”, https://arxiv.org/abs/1610.02415
9. DeepChem, http://deepchem.io
10. ”In-Place Associative Computing”, https://www.gsitechnology.com/APU
11. Biovia Pipeline Pilot, http://www.3dsbiovia.com/products/collaborative-science/biovia-pipeline-pilot/

